Replication: GCRMA (background, normalization & summarization)
Author: Mark Robinson and Henrik Bengtsson
Created: 2009-05-17
Last modified: 2010-02-05
Description
This test verifies that aroma.affymetrix can reproduce the GCRMA (Wu, Irizarry, Gentleman, Murillo, and Spencer, 2004) chip-effect estimates as estimated by the gcrma package.
Data set
rawData/
Affymetrix-HeartBrain/
HG-U133_Plus_2/
u1332plus_ivt_cerebellum_A.CEL [13555904 bytes]
u1332plus_ivt_cerebellum_B.CEL [13550687 bytes]
u1332plus_ivt_cerebellum_C.CEL [13551860 bytes]
u1332plus_ivt_heart_A.CEL [13554731 bytes]
u1332plus_ivt_heart_B.CEL [13553255 bytes]
u1332plus_ivt_heart_C.CEL [13551203 bytes]
Source: Affymetrix Tissue samples, 2007. See Affymetrix data sets for chip type HG-U133_Plus_2.
Script
GCRMA estimates by aroma.affymetrix
library("aroma.affymetrix")
cs <- AffymetrixCelSet$byName("Affymetrix-HeartBrain", chipType="HG-U133_Plus_2")
# GCRMA background correction
bc <- GcRmaBackgroundCorrection(cs)
csB <- process(bc)
# RMA quantile normalization
qn <- QuantileNormalization(csB, typesToUpdate="pm")
csN <- process(qn)
# RMA probe summarization
plm <- RmaPlm(csN, flavor="oligo")
fit(plm)
# Extract chip effects on the log2 scale
ces <- getChipEffectSet(plm)
theta <- extractMatrix(ces)
rownames(theta) <- getUnitNames(cdf)
theta <- log2(theta)
GCRMA estimate by gcrma
library("gcrma"); # gcrma()
raw <- ReadAffy(filenames=getPathnames(cs))
es <- gcrma(raw, verbose=TRUE)
theta0 <- exprs(es)
Results
# Reorder the aroma.affymetrix estimates
o <- match(rownames(theta0), rownames(theta))
theta <- theta[o,]
# (a) Assert correlations
cors <- sapply(1:ncol(theta), FUN=function(cc) cor(theta[,cc],
theta0[,cc]))
print(cors)
print(range(cors))
stopifnot(all(cors > 0.99995))
# (b) Assert differences
e <- (theta - theta0)
stopifnot(mean(as.vector(e\^2)) < 1e-3)
stopifnot(sd(as.vector(e\^2)) < 1e-3)
stopifnot(quantile(abs(e), 0.99) < 0.05)
stopifnot(max(abs(e)) < 0.085)
# (c) Visual comparison
layout(matrix(1:9, ncol=3, byrow=TRUE))
xlab <- expression(log[2](theta[gcrma]))
ylab <- expression(log[2](theta[aroma.affymetrix]))
for (kk in seq(length=ncol(theta))) {
main <- colnames(theta)[kk]
plot(theta0[,kk], theta[,kk], pch=".", xlab=xlab, ylab=ylab,
main=main)
abline(0,1, col="blue")
}
xlab <- expression(log[2](theta[aroma.affymetrix]/theta[gcrma]))
plotDensity(e, xlab=xlab);
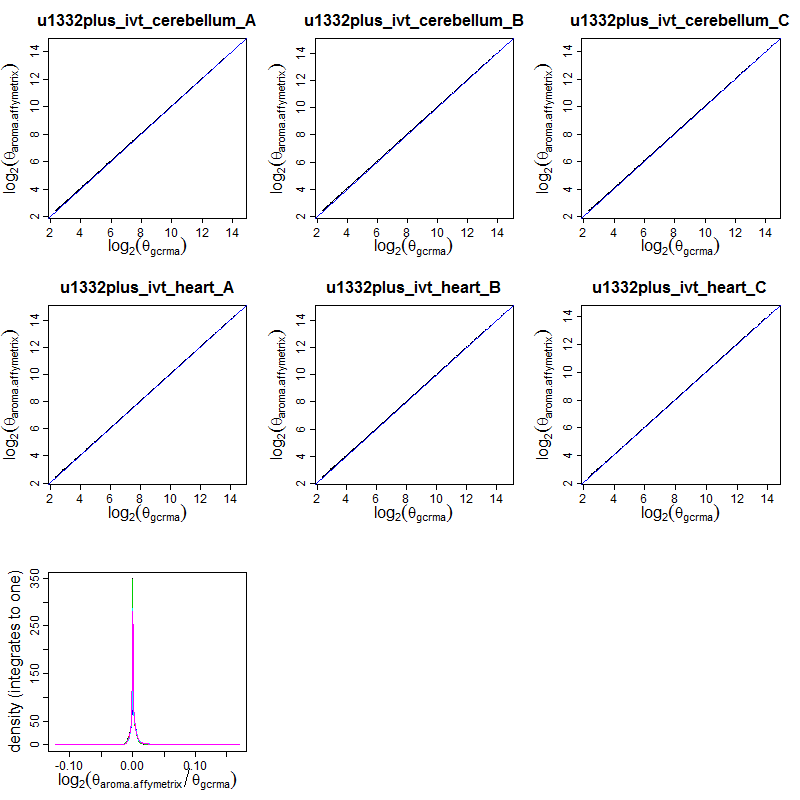
Figure: (Top six panels): Scatter plots comparing the chip-effect estimates (on the log2 scale) from aroma.affymetrix with the ones from gcrma. (Bottom panel): The density of the log2-ratios between aroma.affymetrix and gcrma chip-effect estimates for the six arrays.
References
[1] Z. Wu, R. Irizarry, R. Gentleman, et al. A Model Based Background Adjustment for Oligonucleotide Expression Arrays. Johns Hopkins University Dept. of Biostatistics Working Paper Series 1001. Berkeley Electronic Press, Jul. 2004. URL: http://ideas.repec.org/p/bep/jhubio/1001.html.
Appendix
Session information
R version 2.10.1 Patched (2010-01-12 r50990)
i386-pc-mingw32
locale:
[1] LC_COLLATE=English_United States.1252
[2] LC_CTYPE=English_United States.1252
[3] LC_MONETARY=English_United States.1252
[4] LC_NUMERIC=C
[5] LC_TIME=English_United States.1252
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] hgu133plus2probe_2.5.0 AnnotationDbi_1.8.1
oligoClasses_1.8.0
[4] hgu133plus2cdf_2.5.0 preprocessCore_1.8.0 gcrma_2.18.1
[7] Biobase_2.6.1 aroma.affymetrix_1.4.3 aroma.apd_0.1.7
[10] affxparser_1.18.0 R.huge_0.2.0 aroma.core_1.4.4
[13] aroma.light_1.15.1 matrixStats_0.1.9 R.rsp_0.3.6
[16] R.filesets_0.7.4 digest_0.4.2 R.cache_0.2.0
[22] oligo_1.10.0 affyPLM_1.22.0 affy_1.24.2
[25] R.methodsS3_1.1.0
loaded via a namespace (and not attached):
[1] affyio_1.14.0 Biostrings_2.14.11 DBI_0.2-4
IRanges_1.4.11
[5] RSQLite_0.7-1 splines_2.10.1 tools_2.10.1i386-pc-mingw32
Redundancy test script
The above script is part of redundancy test suite executed at every new release:
path <- system.file("testScripts/replication/chipTypes/HG-U133_Plus_2", package="aroma.affymetrix")
filename <- "12.doGCRMA_vs_gcrma.R"
pathname <- file.path(path, filename)
source(pathname)